

Posts Tagged ‘“Boston product liability lawyers”’
General Motors’ $35 Million Fine Starts Years of Investigations, Wrongful Death Lawsuits
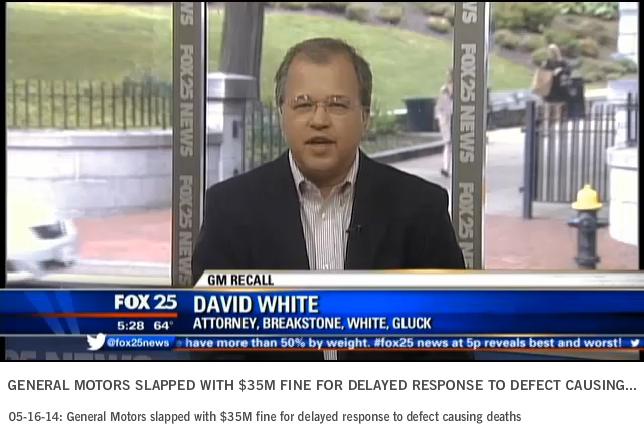
General Motors (GM) has been fined $35 million for waiting a decade to recall vehicles with faulty ignition switches. The defects have now been linked to 13 deaths.
Attorney David White of Breakstone, White & Gluck appeared on Fox 25 TV in Boston last week to discuss the fine, GM’s recent bankruptcy proceedings and his thoughts about handling recall notices.
This is an important topic because we have seen many auto recalls in recent years, but 2014 may set the record, according to this Los Angeles Times article.
White said the fine is the first step in a long process ahead.
“Really this is just a slap on the wrist for the corporation,” White said. “It’s a civil fine. It’s the maximum civil fine that they could be exposed to at this point, but GM is looking at years of investigation and probably… maybe even billions of dollars of fines down the road.”
The federal government is sending a strong message that companies need to act within 5 days of learning of safety defects, as required by law, White said.
“Hopefully other automakers get this message and they tune into the need for greater safety, greater attention to safety,” he said. “When they do find a defect, they come right out and say here’s our defect, here’s our concern so consumers can get notice of it promptly and get it fixed.”
Read More
Household Safety: Check Old Appliances Before Winter
 Now, as we head into winter, is a good time to test your home’s smoke alarms, check your appliances and inspect your electrical outlets and cords.
Now, as we head into winter, is a good time to test your home’s smoke alarms, check your appliances and inspect your electrical outlets and cords.
First, a good resource is the Consumer Product Safety Commission (CPSC) website, where you can search for recall news about products you may own. Recently, there have been several recalls involving products posing serious fire hazards.
One example is Schneider Electric IT Corp. recalled 15 million APC Surge Arrest surge protectors in early October. This followed 700 reports of property damage, including $916,000 in fire damage to a home and $750,000 to a medical facility. Another 13 reports were injuries, including smoke inhalation and contact burns. Another example is Gree Electric Dehumidifiers, which recalled 2.2 million dehumidifiers in the U.S. last month, after its products caused 46 fires and $2.15 million in property damage.
You can review the CPSC website to make sure you have no recalled products in your home. You can also take a look around your home for faulty cords or products.
Preventing Home Fires in the Winter
During a typical year, there are over 26,000 home electrical fires in this country, according to the U.S. Fire Administration. December and January see the most electrical fires. We share a few tips for preventing these fires:
1) Check your smoke alarms before the season.
2) Regularly check your electrical appliances and wiring. Replace any old or damaged cords; do not try to repair them.
3) Replace any appliance you feel may not work properly. If you do not want to replace it, call a repair service or visit the store where you purchased the product. Check electric space heaters every year as a rule.
4) Buy appliances which have the label of a recognized testing laboratory, such as UL.
5) Avoid using extension cords.
6) Use only surge protectors or power strips that have internal overload protection and have been tested by a national laboratory.
7) Keep clothes and flammables at least three feet away from all portable electric space heaters.
8) Use light bulbs that match recommended wattages for lamps.
9) Bring in an electrician if you are experiencing flickering lights or other problems.
Related:
Electrical Home Fire Safety, U.S. Fire Administration
Schneider Electric Recalls APC Surge Protectors Due to Fire Hazard, Consumer Product Safety Commission.
Read More
Recalled Heart Device and Similar Model Under FDA Watch
 The Food and Drug Administration (FDA) has ordered St. Jude’s Medical to expand studies into Durata defibrillator leads, saying the heart devices are sufficiently similar to Riata leads, which were recalled last year. The agency is also seeking new data on Riata.
The Food and Drug Administration (FDA) has ordered St. Jude’s Medical to expand studies into Durata defibrillator leads, saying the heart devices are sufficiently similar to Riata leads, which were recalled last year. The agency is also seeking new data on Riata.
Defibrillators are implanted in patients with abnormal heart rhythms to regulate the heart beat. The medical devices are connected to the heart with leads. Riata was recalled because in some cases, wires were breaking through the insulation, which can potentially lead to a life-threatening abnormal heart rhythm.
A recent St. Jude study of 700 Riata patients found the defective wire broke through the insulation in about 19 percent of cases. A study led by a Minneapolis Heart Institute cardiologist has linked Riata leads to 20 deaths.
St. Jude, of St. Paul, Minnesota, pulled the defective Riata leads off the market in December 2010 and the FDA issued a medical device recall in late 2011. A 2011 count found 79,000 patients were still implanted with the Riata leads.
Responding to the FDA order, St. Jude said it is already collecting data on both Riata and Durata. The company stated the medical devices are different in design and a new coating meant to protect the insulation.
The FDA’s orders include routine X-rays for patients enrolled in post-market Durata studies to identify any insulation problems. St. Jude must also perform X-rays in a three-year post-market study on the defective Riata leads. The goal is to detect extruded leads which are floating loose in the heart.
Surgery to remove extruded leads can be dangerous. St. Jude says many patients live with the loose wires and continue to function normally. The FDA has advised doctors to closely monitor patients.
Related:
- FDA Orders Review of Heart Devices, The Wall Street Journal.
- Riata patients should get X-rays, FDA says, Star Tribune.
- Doctors grapple with FDA advice for troubled heart wire, Fox News.com.
Surgical Mesh Maker to Phase Out Defective Medical Device
Johnson & Johnson, one of the largest manufacturers of surgical mesh, has announced plans to stop selling the medical implants, as it faces hundreds of lawsuits from injured women and increasing oversight from the Food and Drug Administration (FDA).
Surgical mesh is used to strengthen the pelvic wall in cases of pelvic organ prolapse (POP) and stress urinary incontinence (SUI). Last year, approximately 75,000 women had a transvaginal procedure using surgical mesh.
Johnson & Johnson disclosed it will phase out surgical mesh in a recent letter sent to New Jersey and West Virginia judges overseeing patient lawsuits against them. The company said it plans to discontinue four mesh products over the next three to nine months.
On July 13, 2011, the FDA issued a public safety advisory stating traditional POP repair with mesh has no advantage over traditional non-mesh repair. Between 2005 and 2010, more than 3,800 women had reported complications and injuries following transvaginal mesh surgery, according to the FDA.
Many more women have stepped forward since the FDA advisory, many of whom have suffered painful, long-term injuries. Some injuries cannot be corrected through surgery. Hundreds of women have filed lawsuits against the manufacturers, which include Johnson & Johnson and Boston Scientific.
In September 2011, an FDA panel heard arguments from advocacy groups, surgeons and consumer organizations. Many wanted to see a surgical mesh recall, others wanted the FDA to implement more oversight. Some sought a reclassification while others said the surgical mesh problem made another case for overhauling the 510(k) system, which allows manufacturers to sell new products without a new review so long as they are “substantially equivalent” to an already legally marketed device.
The FDA did not require a surgical mesh recall. But in January 2012, it ordered Johnson & Johnson and the other companies to conduct studies to track surgical mesh complication rates over time. Johnson & Johnson has been collecting data to comply, but said it expects the FDA will waive the requirement.
Complaints from transvaginal surgical included mesh erosion with in the body, bleeding, pain during sexual intercourse, organ perforation, vaginal scarring, muscular and emotional problems. While corrective surgery is an option, it is not always successful.
Some surgeons are now recommending women consider whether they really need POP surgery and if they do, request a procedure without mesh, according to a Consumer Reports article. Click here for a recent article.
Related:
- FDA Safety Communication: UPDATE on Serious Complications Associated with Transvaginal Placement of Surgical Mesh for Pelvic Organ Prolapse, July 13, 2011.
- What to Know If You Have Been Injured By Surgical Mesh or Transvaginal Mesh.
- Urogynecologic Surgical Mesh Implants, Food and Drug Administration
Defective Bikes and Equipment List for Cyclists
 Each spring, cyclists throughout Massachusetts say goodbye to winter and put their bikes back on the road. But whether you cycle every day or just occasionally, now is the time to make sure your bike and equipment meets the latest safety regulations.
Each spring, cyclists throughout Massachusetts say goodbye to winter and put their bikes back on the road. But whether you cycle every day or just occasionally, now is the time to make sure your bike and equipment meets the latest safety regulations.
It is good practice to test and inspect key parts of your bike, such as the quick release wheels, brakes and pedals.Then check with the manufacturer of your bicycle. Look online and see if if offers an owners’ manual. If you have not done so, register your bike so you may receive recall notices.
You can also check the Consumer Product Safety Commission (CPSC) website for recent bicycle recalls. Each year, the CPSC recalls hundreds of thousands of bicycles and parts after receiving reports of defects and injuries.
Here are a few recent bicycle and equipment recalls from the CPSC:
Recalled Bicycles. Some 91,000 Bridgeway Bicycles were recalled in September 2011 because of a defective bicycle chain which can break, causing the rider to lose control and fall. The CPSC received 11 incident reports, including injuries, lacerations and contusions. Read more.
Children Bicycle Seats and Trailers. Two of the largest bicycle-related recalls involve defective children’s equipment. Topeak Babyseat II Bicycle Carrier Seats were recalled in April 2012 after two reports of near amputations and crushed fingers. When a child is lifted out of the seat, their fingers can get caught in a defective hinge mechanism. The product recall affected 30,400 consumers. Read more.
In January 2012, 44,000 Chariot bicycle trailers and 70,000 trailer conversion kits were recalled after 24 incident reports around the world, including three in the U.S. The trailer’s hitch mechanism can crack and break, causing the trailer to detach from the bicycle. Read more.
Helmets. Little Tricky Bicycle Helmets recalled 30,400 bicycle helmets in January 2012. Product testing demonstrated the helmets did not comply with CPSC safety standards for impact resistance. Read more.
Click for a full list of recent bicycle-related recalls from the CPSC.
Read More
Defective Medical Devices Gain Criticism from Consumer Reports
 Consumer Reports is stepping into the debate about the medical device approval process, recommending that the Food and Drug Administration require more rigorous testing to prevent defective medical devices from going to market.
Consumer Reports is stepping into the debate about the medical device approval process, recommending that the Food and Drug Administration require more rigorous testing to prevent defective medical devices from going to market.
The magazine and its advocacy arm Consumers Union wants the FDA to require implants and other medical devices undergo testing to prove they are safe and effective. The FDA began classifying medical devices into three categories in 1976 and stated manufacturers would be required to show clinical data before approval of Class III products, the most at-risk category.
But the FDA routinely clears new medical devices under a process known as 510, in which manufacturers are required to bypass clinical testing if they can show a device is “substantially equivalent” to another device already on the market.
Consumers Reports is calling on Congress to require testing as part of the FDA’s approval process for medical devices. Next, it wants the practice of “grandfathering” high-risk implants stopped. Finally, the organizations seek an improvement to the system for notifying patients of medical device failures.
Currently, the system largely relies on physicians who are supposed to notify patients, but this is a problem when doctors stop practicing.
Without changes to the system, Consumer Reports said patients cannot properly protect themselves.
The magazine highlighted three types of defective medical devices which have caused injuries in recent years:
Surgical Mesh: This device was approved several years ago based on its relationship to a product used in the 1950s, even though the two products were inserted differently and treated different areas of the body. The FDA refused calls to recall surgical mesh, but in January ordered 33 companies to conduct the first-ever post-market safety studies of the product. The FDA is also considering reclassifying surgical mesh into a Class III category.
The Consumer Reports article shares the story of a 54-year-old woman who has undergone eight surgeries to correct her transvaginal mesh complications.
Artificial All-Metal Hips: DePuy Orthopedics recalled its ASR XL all-metal hip implant in 2010 after the FDA received about 400 complaints in two years from patients. The two metal parts were rubbing against each other, breaking and spreading metal particles into the blood stream. Injury reports about all-metal hip implants grew after that, with the FDA receiving more than 5,000 reports about hip implants in the first six months of 2011, according to a New York Times article. DePuy hip implants was estimated to account for 75 percent of those injury reports.
Related:
- Consumer Reports Investigates: Dangerous Medical Devices
- Protect Yourself Against Medical Device Injuries
Recalled Birth Control Pills: Lo/Ovral-28, Norgestrel and Ethinyl Estradiol
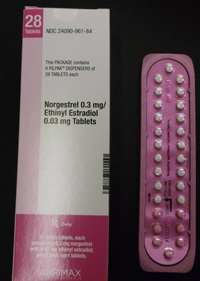 Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
Pfizer Inc. has voluntarily recalled certain lots of birth control pills which may contain ingredient errors or out-of-sequence packaging which could have exposed women to a risk for unintended pregnancy.
In January, Pfizer recalled 14 lots of Lo/Ovral-28 (norgestrel and ethinyl estradiol) Tablets and 14 lots of Norgestrel and Ethinyl Estradiol Tablets (generic) for customers in the U.S. market. The defective pills were distributed to warehouses, clinics and retail pharmacies nationwide.
An investigation by Pfizer found that some blister packs may contain an inexact count of inert or active ingredient tablets and that the tablets may be out of sequence. Pfizer recalled the tablets on January 31, 2012, with knowledge of the Food and Drug Administration (FDA). Pfizer said the error has been corrected.
The tablets were manufactured and packaged by Pfizer Inc., commercialized by Akrimax Rx Products and labeled under the Akrimax Pharmaceuticals brand. The medicine is packaged in blister packs of 21 tablets of active ingredients and seven tablets of inert ingredients. Click the link below for packaging numbers involved in the recall.
The product liability lawyers at Breakstone, White & Gluck are reviewing cases for women who have taken defective lots of these birth control pills and have experienced or are experiencing an unplanned pregnancy. Contact us today at 800-3791244 or 617-723-7676 or use our contact form. Read More
Defective Medication Under Scrutiny After Supreme Court Ruling
A recent Supreme Court ruling is limiting court actions by injured patients who have filed claims against manufacturers of generic drugs.
The ruling was issued last year and said generic drugmakers do not have control over their labels and therefore cannot be sued for failing to alert the public. Under the 1984 Hatch-Waxman Act, generic drugmakers were not required to undergo the Food and Drug Administration’s (FDA) lengthy approval process if they could prove the generic drug was equivalent to the brand-name medicine.
In most cases, the Henry-Waxman Act requires generic manufacturers use the same labels as brand-name drugs, with dosing instructions and risks for injury. For this reason, judges have started to dismiss many product liability lawsuits against generic manufacturers while allowing those against brand-name drugs to move ahead.
In a March 20, 2012 article, The New York Times reported that a woman who had received the brand name for an anti-nausea medication had suffered gangrene – or a condition that results in dead or weakening body tissue. She sued the manufacturer Wyeth and won $6.8 million.
Another woman took the generic version of the defective drug, known as promethazine, and had to have her arm and forearm amputated because of complications from gangrene. Her case was dismissed last fall following the Supreme Court ruling.
The Supreme Court ruling comes as Americans are increasingly turning to generic medicines. As prices skyrocket and the economy struggles, many health insurance companies are requiring generics be filled before brand-name drugs. Doctors are required to show medical needs for the brand name over generic.
As a result, nearly 80 percent of prescriptions in the United States are filled generic and most states permit pharmacists to dispense a generic in place of a brand name.
What Can Consumers Do:
Support efforts to change the law. Public Citizen, a consumer advocacy group, has petitioned the FDA to give generic companies greater control over their labels. The move may allow generic drug users to sue. U.S. Rep. Henry A. Waxman, D-California, is also exploring ways to address the issue.
Talk to your doctor. Ask your doctor about the medicine being prescribed, the generic and potential side effects. If you are still concerned about potential injuries, ask your doctor to call your insurance company and request a brand-name.
Research any medication you use. Write down the name of the medicine you are prescribed, the medicine you receive at the pharmacy and research both drugs. Discuss any side effects with your physician.
Consider foregoing insurance. If you are really concerned and can afford the brand-name prescription, consider purchasing it. There are many discount drug programs which may help you reduce your costs. Check with any groups you are affiliated with, including AAA and AARP.
Contact your health insurance company. If the company has required you to use generic medications, ask if it has changed its policy and is now allowing use of brand-name medications.
Related:
Massachusetts Patients’ Bill of Rights and Preventing Medical Errors.
Product Recall: More than 10,000 Fuji Bicycles Recalled
 The U.S. Consumer Product Safety Commission announced the recall this week of more than 10,000 defective bicycles after reports the product’s frame was breaking.
The U.S. Consumer Product Safety Commission announced the recall this week of more than 10,000 defective bicycles after reports the product’s frame was breaking.
Fuji Saratoga Women’s Bicycles recalled about 10,500 bicycles sold nationwide from November 2007 through December 2011. The bicycles were recalled after the company became aware of 12 reports of bicycle frames breaking. There were two injuries reported, including a head laceration requiring 20 stitches.
The product’s defect is its frame was breaking in the center of the downtube during use, causing bicyclists to lose control and fall. Bicyclists are instructed to stop riding and seek a replacement bike frame.
About the recalled Fuji women’s cruisers bicycles:
- 2008 to 2010 models of Saratoga 1.0, Saratoga 2.0, Saratoga 3.0 and Saratoga 4.0. The model type will be printed on the bike.
- The bikes come in various colors.
- The bikes will have the words “Fuji” and “Saratoga” alone or “Saratoga” printed on the frame.
- Serial numbers beginning with ICFJ7, ICFJ8, ICFJ9, ICFJ10 and ICFJ11. The serial number is located on the bottom of the frame near the crank.
The defective products were imported by Advanced Sports, Inc. of Philadelphia and manufactured in China. They were sold at specialty bike shops.
Customers are instructed to obtain a free replacement frame. They can contact Advanced Sports Inc. toll-free at 888-286-6263 between 8 a.m. and 4:30 p.m. Monday through Friday or visit www.fujibikes.com. They can also return the bike to any authorized Fuji Bicycle dealer for the free part.
Click here for more information on this recall.
In a smaller recall, the Mountain Bicycle Handlebar Stem has been recalled in the U.S. and Canada. Some 213 units were recalled in the U.S. and 83 in Canada by the importer, Shimano American Corp. of Irvine, Calif. The defective bicycles were sold at REI stores nationwide from October 2009 to November 2010 for about $120.
The bicycles were recalled because the bolt holding the front plate of the stem to the stem body can be pulled out of the threads while the bike is being ridden, causing the rider to fall. There has been one report of a rider falling and sustaining torso and arm injuries. Click here for more information on this recall.
The Boston product liability lawyers at Breakstone, White & Gluck have over 80 years combined experience handling complex cases involving serious personal injuries, wrongful death and defective products. We have obtained clients compensation in cases involving defective motor vehicles, recalled medical devices and dangerous pharmaceuticals.
If you have been injured, it is important to learn your rights and how long you may have to file a claim. For a free legal consultation, contact us today at 800-379-1244 or 617-723-7676 or use our contact form.
Recalled Coffee Makers Burn Nearly 40 People
 More than 1.7 million coffee makers have been recalled after reports some machines have sprayed hot liquid, leaving 37 people with second-degree burns.
More than 1.7 million coffee makers have been recalled after reports some machines have sprayed hot liquid, leaving 37 people with second-degree burns.
The Tassimo Single-Cup Brewers were recalled Feb. 9 by BSH Home Appliances Corp. of Irvine, California. Some 835,000 machines were recalled in the United States and 900,000 were recalled in Canada. The California manufacturer recalled the defective product voluntarily along with the Consumer Product Safety Commission (CPSC) and Health Canada.
The brewers are defective because they can burst and spray hot liquid and coffee grounds or tea leaves onto consumers. There were a total of 140 reports of the brewers spraying hot liquid. Among the 37 second-degree burns was a 10-year-old Minnesota girl who suffered serious facial and neck burns which required her to be hospitalized.
The defective coffee makers carried the brand names of Bosch and Tassimo Professional Brewers. The Bosch brewers were sold in several colors to consumers between the dates of June 2008 and February 2012 for between $100 and $250. The Tassimo Professional was sold on in black, directly to hotels and food service providers. The brewers were manufactured in Slovenia and China.
Consumers are advised to stop using the recalled coffee makers immediately and contact the firm to order a free replacement T Disc holder part to fix the mechanism. This is the part of the machine that holds the single serve coffee cup.
Consumers can visit www.tassimodirect.com/safetyrecall for the full list of recalled models and to request a replacement part. They can also call the firm toll-free at 866-918-8763.
Click here to read the recall notice from the CPSC.
Read More